




01、研究背景
镁合金因其轻质特性,在汽车和航天工业领域具有巨大的应用潜力,可显著提高能源效率,减少二氧化碳排放。然而,镁合金较低的强度阻碍了它们规模化应用。提高镁合金强度最常用的方法是变形,比如挤压,但同时也不可避免地会在合金中引入织构缺陷。挤压镁合金中,<1-100>丝织构占据主导,从而导致在特定加载方向下的变形过程中,如沿着挤压方向压缩,{10-12}孪生将成为主要的塑性变形机制。
孪晶界在微观组织调控和力学性能方面具有多元化作用,因此大量的研究人员都致力于揭示镁合金中孪晶的结构及其形成机理。前几年,朱玉满等人利用原子分辨率的高角环形暗场扫描透射电镜,细致观察了压缩变形量达到27%的Mg-Gd合金中的多种倾斜界面的结构和元素偏聚;揭示了多种非对称界面被,如0°/86°, 26°/60°, 16°/36°, 10°/54°, 20°/60°, 0°/56°和12°/44°。但至今为止,大部分非对称界面的形成机理并不清楚。近期研究显示,共格孪晶界上的非对称界面是研究非对称界面形成机理以及孪晶在塑性变形中作用的关键。众所周知,室温下{10-12}孪生是继基面滑移后最容易激活的变形方式,而孪晶界的迁移对于镁合金塑性变形起着关键作用。从晶体学上,{10-12}面不是平坦的,本质上呈波纹状,由两个不重合的子平面组成。因此,在{10-12}孪晶界上总是会出现基面-柱面匹配界面(BP界面)或者其他非对称界面。BP界面是{10-12}孪晶界面最常见的,也被认为是不可动的;但{10-12}孪晶界上其他非对称界面很少被观察到,更别说它们的形成方式。因此,揭示{10-12}孪晶界上的特殊非对称界面对于更好地理解孪晶与位错的反应致关重要。
最近,中国科学院长春应用化学研究所杨强研究员和邱鑫研究员,联合吉林大学陈鹏教授和长春理工大学吕术慧教授,针对镁合金中非对称界面的形成机理很难通过传统理论模型来揭示的难题,结合原子级HAADF-STEM表征、分子动力学模拟和第一性原理计算,对镁合金中{10-12}孪晶界上的特殊非对称台阶和界面的原子结构和形成机理进行了研究。镁基体中的<a60>位错与{10-12}共格孪晶界反应会形成一个∼61°/25°非对称倾斜界台阶,大量的<a60>位错同时与{10-12}孪晶界反应则会形成长度超过30 nm的非对称界面;合金元素的偏聚位点与界面位错和界面间隙密切相关,且合金元素偏聚有利于降低体系总能。该研究可用于指导镁合金的成分设计,通过降低孪晶界上非对称界面形成能从而提高镁合金的塑性和成形性。
02、图文导读
文章首先通过传统热挤压工艺制备了Mg−12Gd−1.5Zn(GZ122)合金,然后室温下沿挤压方向压缩~7%;然后通过双球差校正扫描透射电子显微镜观察{10-12}孪晶边界的结构,并通过四探头高分辨EDS对特殊非对称界面的元素偏聚进行了分析;结合实验结果,作者构建了<a>位错与{10-12}孪晶界的反应模型,并利用LAMMPS进行了分子动力学模拟,同时也构建了18.1°/62.3°界面模型(182个原子),利用VASP 5.2进行了DFT(Density functional theory)计算。拉伸和压缩试验结果表明该合金在室温下压缩塑性显著高于拉伸,表明孪生对镁合金塑性的贡献非常大,相应的EBSD和TEM研究表明该合金中最主要的孪晶是{10-12}。通过细致观察{10-12}孪晶的界面,我们发现部分台阶不是常规的BP台阶,而是一种20.5°/65.8°非对称台阶,如图1a所示;相应的拓扑分析结构表面,这种非对称台阶是由基体中的<a60>位错与{10-12}孪晶界反应形成的(图1b)。同样,如果孪晶中的<a60>位错与{10-12}孪晶界反应,也可以形成非对称台阶,如24.8°/59.8°界面(图1c和d)。
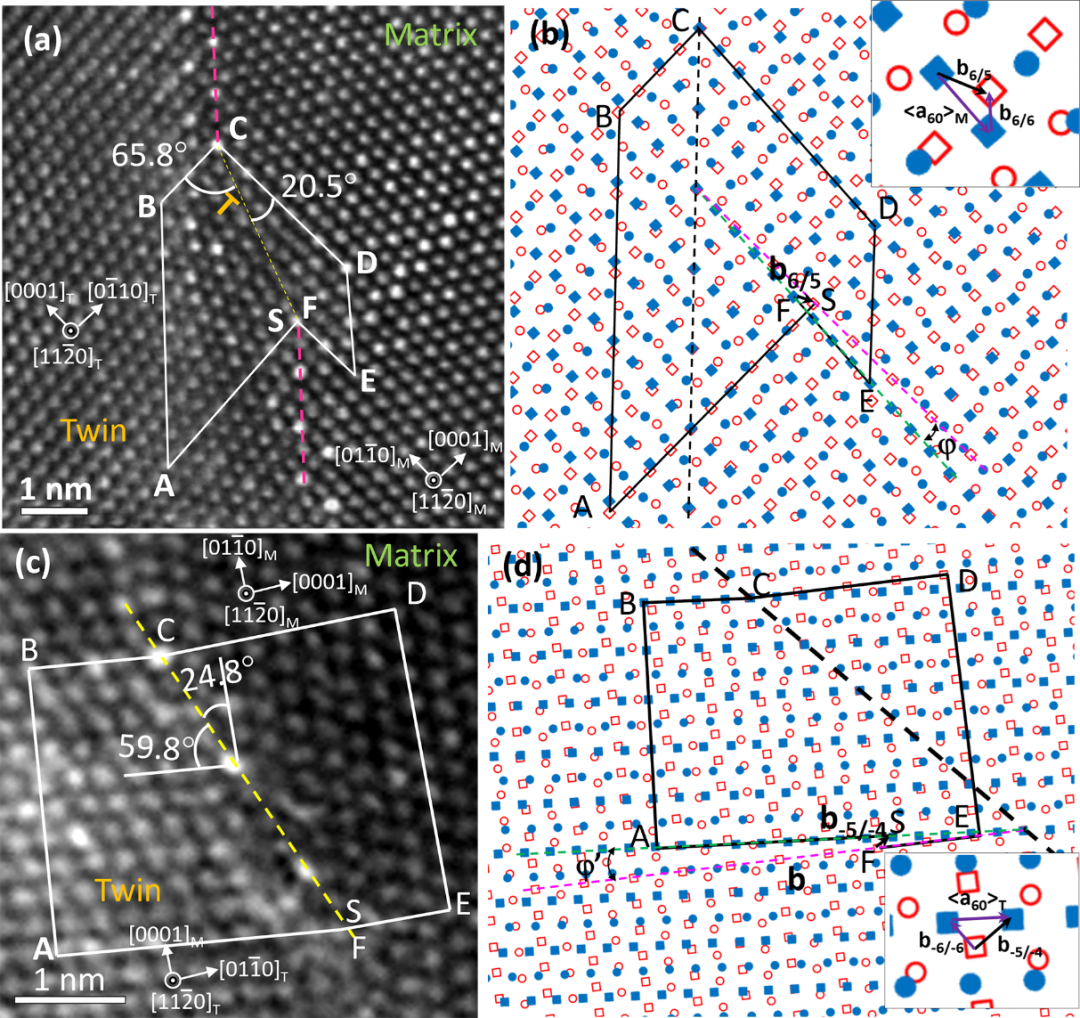
图1 孪晶界上非对称台阶的原子分辨率HAADF-STEM照片及相应的界面位错拓普分析
在{10-12}孪晶界上,作者还发现了长度超过30 nm的18.1°/62.3°非对称界面(图2a),界面上有密集的等间距分布的位错,位错间距为2或3个基面高度(图2b)。通过放大图可以发现,这些界面位错的伯氏矢量为b4/3或b6/5(图2c和d)。通过低倍图(图2e)发现在非对称界面下方还存在一个由位错塞积形成的亚晶界,角度为4.3°。细致对比发现,下方亚晶界上位错的数量与非对称界面上的位错数量几乎相等,只是亚晶界上位错分布并不是等间距的。同时,合金元素Gd和Zn会偏聚到位错核心的拉应力区,有时也会形成βF’- βF’ 结构。
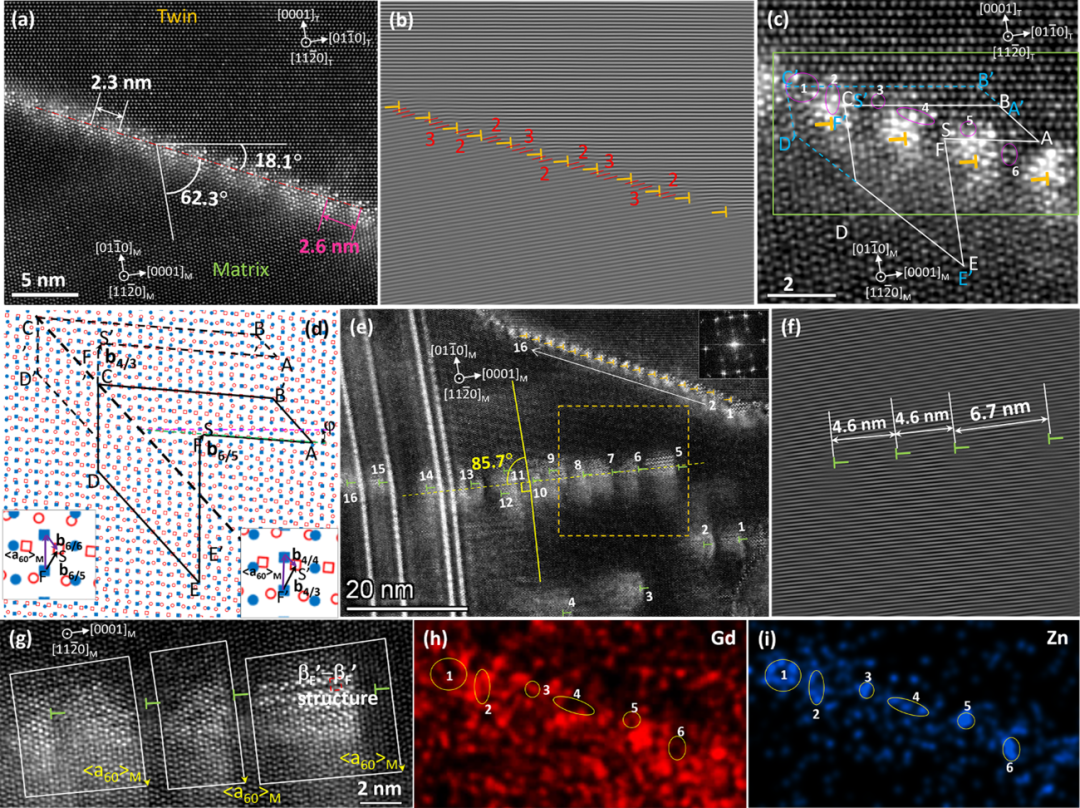
图2 (a-c)非对称界面的原子分辨率HAADF-STEM图及(d)界面位错拓普分析,(e-g)非对称界面周围基体中的位错分布及伯氏矢量分析,以及非对称界面的EDS面扫图谱:(h)Gd元素和(i)Zn元素。
基于上述实验结果,作者构建了基体中<a60>位错与{10-12}孪晶界反应的模型并进行了分子动力学(MD)模拟。当位错前端达到{10-12}孪晶界时,首先会形成一个短的BP台阶(图3a);随着孪晶界上移,在它的左侧孪晶界上会形成好几个短的BP台阶(图3b),然后这些短的BP台阶再消失,在靠近最先形成的BP台阶处形成一个短的非对称倾斜台阶(图3c);当<a60>位错完全移动到最先形成的短BP台阶上时,一个明显的非对称台阶也就形成了(图3d)。通过对非对称台阶的放大图分析可以发现,该非对称台阶为61°/25°(图3e),界面位错位b6/5,这与实验结果非常吻合。众所周知,在孪晶尖端通常会有大量的BP台阶,同时变形过程中也会形成大量的亚晶界,当亚晶界上的一个位错与孪晶界上的一个BP台阶反应时会形成一个短的61°/25°非对称台阶(图3g和h),同时BP台阶向右移动并与新到的位错反应,从而实现非对称台阶的长大(图3i);当一排位错与BP反应结束后,一条非对称界面形成,界面取向偏离孪晶界~21.1°(图3j)。上述原子级HAADF-STEM表征结果表明在这条非对称界面上也形成了元素近周期性偏聚,作者构建了18.1°/62.3°界面模型,如图4a所示;该模型同时包含两个位错核心,间距为2或3层基面间距。在界面附近作者选取了54个位点,Gd和Zn分别占据这些位点体系能量都会降低,而部分位点占据出现了明显的能量低谷(图4b),表明这些位点更有利于Gd和Zn的偏聚。DFT计算的Gd和Zn偏聚位点与实验观察到的结果较为吻合(图4c-f)。在界面上,Gd元素倾向于偏聚到界面位错核心,Zn更倾向偏聚到小的界面间隙附近;合金元素在非对称界面上的偏聚有利于体系能量的降低。
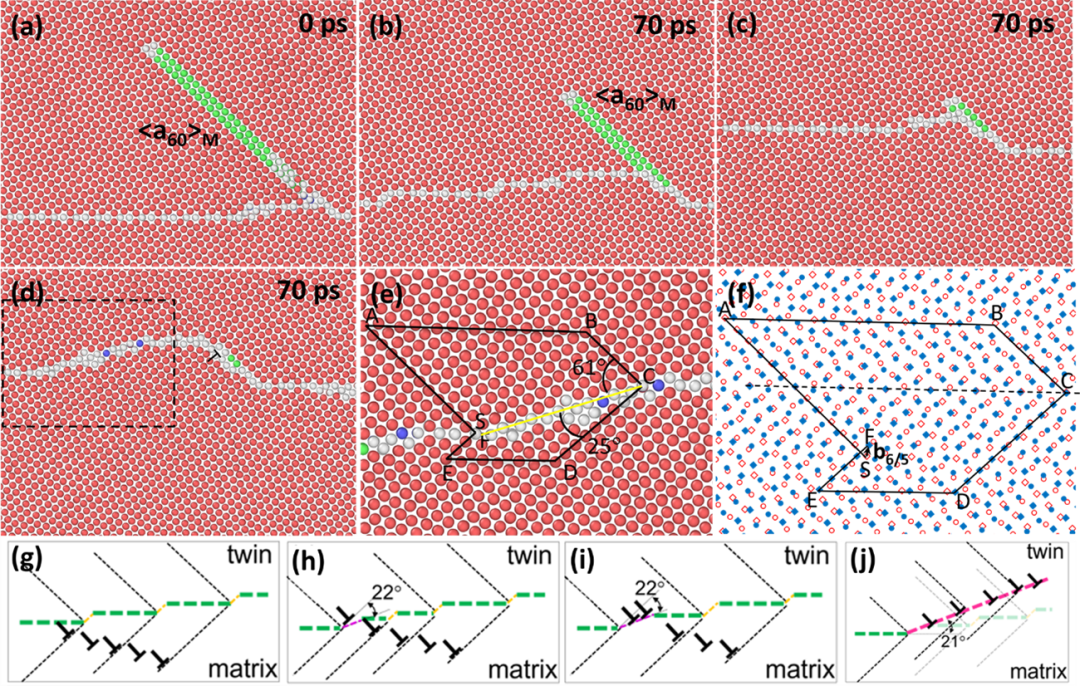
图3 (a-d)MD模拟镁基体中<a60>位错与{10-12}孪晶界反应过程以及(e,f)形成的非对称界面的位错分析,(g-j)非对称界面形成过程示意图。
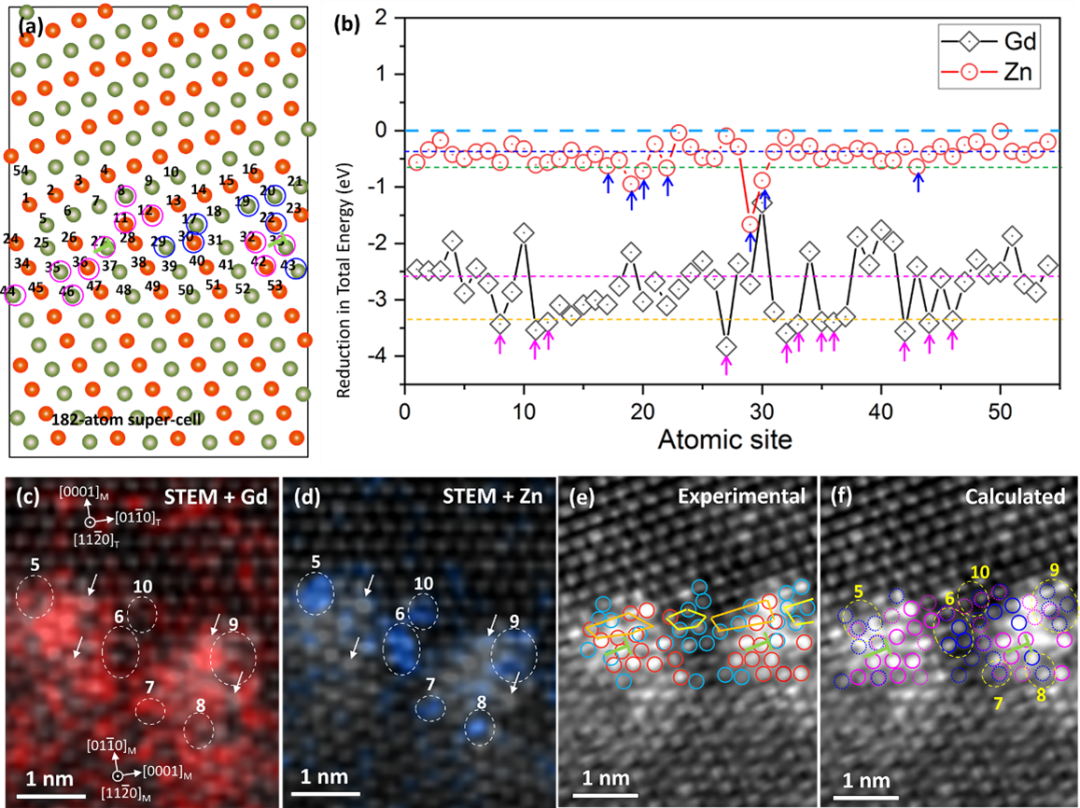
图4 Gd和Zn再非对称界面上偏聚体系能量变化DFT计算结果及Gd和Zn计算偏聚位点与实验偏聚位点的比较
03、结论与展望
综上所述,镁基体中<a60>位错与{10-12}孪晶界反应除了可形成常见的BP台阶外,还可以形成∼61°/∼25°非对称倾斜台阶,形成的界面位错位b6/5。当一定量的<a60>位错与{10-12}孪晶界连续反应时则会形成一段非对称界面,长度可超过30 nm。在非对称界面上,Gd元素更倾向于偏聚到界面上堆积的<a>位错核心,而Zn元素更倾向于偏聚到小界面间隙附近;合金元素的偏聚有利于体系能量降低,进一步稳定了非对称界面结构。该研究发现了一种新的{10-12}界面结构,也提供了一种新的孪晶界迁移机制,这对于提高镁合金塑性的镁成分和工艺设计具有指导意义。
04、文章信息
该文章发表在《Journal of Magnesium and Alloys》2025年第13卷第2期:[1] Qiang Yang, Shuhui Lv*, Peng Chen*, Zefeng Xie, Shuo Zhou, Xin Qiu*. Formation and solute segregation for an asymmetric tilt boundary on {10-12} twin boundaries [J]. Journal of Magnesium and Alloys, 2025, 13(2):583-591.
05、下载链接

扫描二维码下载文章!
06、中文摘要
传统孪晶界上的非对称界面是揭示孪晶在金属材料中协调塑性变形机制的关键。但是大部分非对称界面的形成方式和机理很难通过传统理论模型来揭示,且非对称界面上合金元素偏聚也非常复杂。本文利用双球差校正-高角环形暗场扫描透射电镜揭示镁合金中拉伸孪晶{10-12}界面上的特殊非对称界面的结构,在通过分子动力学模拟和第一性原理计算,揭示非对称界面的形成机理及元素偏聚机理。研究结果表明,镁基体中<a60>位错与{10-12}孪晶界反应除了可形成常见的BP台阶外,还可以形成∼61°/∼25°非对称倾斜台阶,并最终长大成非对称界面,长度可超过30 nm。在非对称界面上,Gd元素更倾向于偏聚到界面上堆积的<a>位错核心,而Zn元素更倾向于偏聚到小界面间隙附近;合金元素的偏聚有利于体系能量降低,进一步稳定了非对称界面结构。
07、英文摘要
Asymmetric tilt boundaries on conventional twin boundaries (TBs) are significant for understanding the role of twins on coordinating plastic deformation in many metallic alloys. However, the formation modes of many asymmetric tilt boundaries are hard to be accounted for based on traditional theoretical models, and the corresponding solute segregation is complex. Herein, atomic structures of a specific asymmetric boundary on {10-12} TBs were reveled using aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), molecular dynamics (MD) and density functional theory (DFT) simulations. Reaction between <a60>M dislocations and the {10-12} TB can generate a ∼61°/25° asymmetric tilt boundary. The segregation of Gd and Zn atoms is closely related to the aggregated <a> dislocations and the interfacial interstices of the asymmetric tilt boundary, which is energetically favorable in reducing the total system energy.
08、作者简介
第一作者/通讯作者简介:
杨强(第一作者),中国科学院长春应用化学研究所研究员,中国科学院青年促进会会员,吉林省高层次人才。稀土镁合金设计开发与产业化应用方向。现担任JMA、RM等七本期刊青年编委;主持了国家及省级基金8项,企业横向多项;以第一/通讯作者在Acta Mater、JMA、JMST等杂志上发表学术论文70多篇,申请发明专利50多项,获省部级和院级等奖励10多项。
吕术慧(通讯作者),长春理工大学教授,博士生导师,吉林省高层次人才。主要研究方向是原子级材料微观组织与性能的研究。现担任JMA期刊青年编委,主持了国家和省级项目多项,以第一/通讯作者发表学术论文40多篇,申请发明专利10余项。
陈鹏(通讯作者),吉林大学教授,先进轻合金团队骨干,入选国家级青年人才计划。于山东大学、中科院金属所、美国内华达大学获本、硕、博学位,美国德克萨斯州农工大学博士后。聚焦轻合金设计与强韧化,在NC、Acta Mater.等发表论文30余篇。
邱鑫(通讯作者),中国科学院长春应用化学研究所研究员,博士生导师,吉林省高层次人才,镁协专家。主持国家级和省级基金、企业横向项目40余项目,获稀土科学技术奖一等奖(排名1),吉林省专利奖金奖(排名第2),吉林省科学技术一等奖(排名第3)等多项奖励。现担任JMA、JER等期刊青年编委;发表学术论文40多篇,申请专利50多项,制定国家标准2项。


