








近日,上海交通大学邹建新团队、德累斯顿工业大学冯新亮/于明浩团队、密歇根大学Richard Laine团队联合在国际期刊《Advanced Materials》发表长篇综述论文“Reassessing Electrolyte Design for Non-Aqueous Magnesium Batteries: Atomistic Structures and Performance Optimization”。该综述系统总结了非水系镁电池电解液的研究进展及其在分子结构、离子输运与界面反应方面的内在关联。文章从含氯与无氯电解液体系出发,深入探讨了溶剂、镁盐及添加剂的设计思路及其对电化学性能的影响,重点分析了镁可逆沉积/剥离过程中的反应机理以及无氯体系在环境友好性与界面稳定性方面的潜在优势。该综述揭示了电解液分子结构对Mg2+溶剂化形态、离子迁移及界面形成过程的调控规律,并提出了“原子结构—溶剂化状态—界面化学—电化学性能”的多尺度关联模型。该工作为高稳定性、高电导率、宽电化学窗口的镁电池电解液设计提供了系统性理论框架与实验指导,对推动镁电池的实用化具有重要意义。
该论文第一作者是:徐昊、贺小倩

研究背景
近年来,随着高效、可持续且高容量储能体系需求的日益迫切,下一代电池技术的研究受到广泛关注。其中,非水系镁电池因镁资源丰富(地壳含量约2.1%)、理论体积容量高(3833 mAh cm−3)、理论质量容量大(2205 mAh g−1),且在充放电过程中不易形成枝晶而具有优异的安全性,被视为锂离子电池的重要替代体系,特别适用于大规模可再生能源储能场景。然而,尽管镁电池在理论性能和安全性方面具备显著优势,其实际应用仍受到电解液体系瓶颈的严重限制。由于Mg2+具有高电荷密度和强库仑相互作用,其在电解液中的溶剂化与迁移过程远比单价Li+复杂,导致离子扩散缓慢、电极界面反应受限及不可逆副反应频发。这些问题导致循环寿命短、库仑效率低和电化学稳定性不足,成为制约镁电池发展的关键障碍。
目前已开发的电解液主要包括含氯与无氯体系。含氯电解液(如APC、MgCl2/AlCl3体系)因Cl−能够促进可逆的镁沉积/剥离,但其强腐蚀性及环境风险限制了实际应用。无氯电解液(如Mg(TFSI)2、Mg(B(hfip)4)2体系)在环境友好性与材料稳定性方面具有潜在优势,但普遍存在镁负极钝化、制备过程复杂及成本高等问题。因此,从原子尺度层面系统理解非水系镁电解液中Mg2+的溶剂化结构、界面反应机制及其与电极的协同关系,并在此基础上实现电解液分子结构与电化学性能的精确调控,成为推动高性能镁电池实现实用化的关键科学问题。
内容表述
1. 镁电池的研究基础与发展趋势
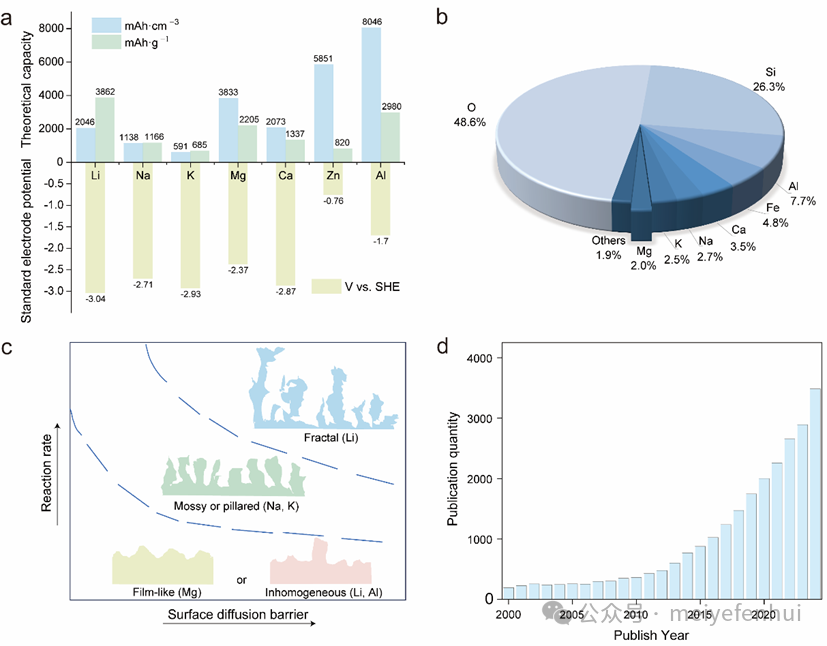
图1. (a)不同金属负极的理论比容量、体积比容量及标准电极电位;(b) 各元素在地壳中的天然丰度;(c) 代表性金属负极的电沉积形貌示意图;
(d) 2000年至2024年间以关键词“Mg battery”在Web of Science数据库中检索得到的镁电池相关论文发表数量统计。
镁负极因其低枝晶形成倾向、资源丰富性及优异的电化学特性,被认为是锂负极的理想替代方案。其兼具较高的体积比容量(3832 mAh cm−3)、较高的质量比容量(2205 mAh g−1)以及较低的标准电极电位(−2.37 V vs. SHE),在保证安全性的同时具备高能量密度潜力。与锂、钠、钾等金属相比,镁在沉积过程中表现出膜状、均匀的电沉积形貌,得益于其较强的金属键结合与较低的成核过电位,有效抑制了枝晶生长,显著提升了电池运行安全性与循环稳定性。相较之下,锂、钠及钾因表面扩散势垒高和成核不均匀而易形成枝晶或苔藓状沉积,铝则表现出不规则形貌。镁致密而平滑的沉积层不仅降低短路风险,还确保了结构完整性和电化学可逆性。此外,镁在地壳中的天然丰度高达2.1%,远超锂(0.0065%),为其在大规模储能系统中的可持续应用奠定了资源基础。伴随研究热度的持续上升,2000年至2024年间“Mg battery”相关论文数量显著增长,充分体现了镁电池在新一代高安全、高能量密度储能技术领域的战略价值与研究前景。
2. 镁电池的电化学反应机制

图2.放电过程中可充镁电池的反应示意图:(a)以Mg2+为主导的反应;(b)双离子(阳离子协同)主导的反应;(c)阴离子参与的正极反应。
镁电池的工作原理基于镁离子在电极间的可逆迁移:放电过程中,金属镁负极经氧化释放Mg2+,其通过电解液迁移至正极并嵌入活性位点,同时电子经外电路输出形成电流;充电时该过程逆转,Mg2+在负极被还原沉积。电解液在其中起到关键作用,不仅需保证Mg2+的高效传输,还必须在循环过程中保持化学与电化学稳定性。非水系镁电池的正极反应主要可分为三类:Mg2+主导型、双离子协同型以及阴离子参与型。Mg2+主导型体系中,Mg2+直接参与电化学嵌入/脱出反应,结构简单但受限于较差的离子扩散动力学。双离子协同型体系中,除Mg2+外,Li+、Na+或K+等阳离子可共同嵌入正极,有助于提高电荷载体数量、改善电化学反应动力学并缓解纯Mg2+体系中常见的结构应变,从而提升循环稳定性和能量效率。然而,该体系对离子浓度控制及电解液设计提出更高要求。阴离子参与型体系则以电解液阴离子(如TFSI−)在正极反应中扮演主导角色,能够在较少Mg2+参与的情况下实现高能量密度,但其循环可逆性和阴离子稳定性仍面临挑战。总体而言,不同反应机制涉及的离子种类差异显著,因此需针对性地设计电解液体系以匹配特定的电荷传输途径与反应特征。
3. 镁负极成核与生长机理
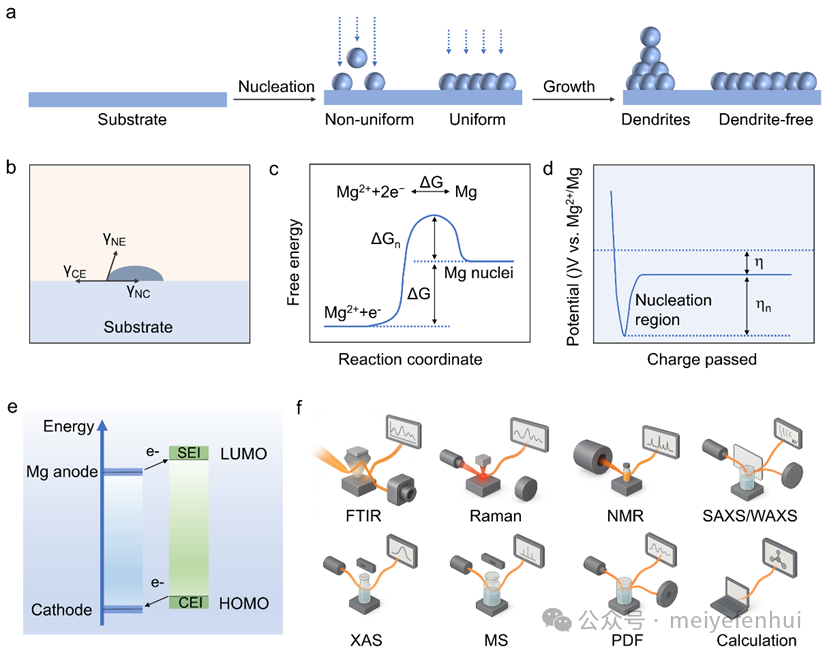
图3. (a)镁在集流体上的成核与生长过程;(b) 基于经典异质成核模型的镁在集流体上的成核示意;(c) 自由能变化的示意图;
(d) 镁恒电流沉积的典型电压曲线;(e) 基于分子轨道能级的SEI形成机制示意;(f) 用于研究电解液组分与分子结构的表征技术。
在镁电池中,镁离子的沉积与剥离过程受其与溶剂分子及阴离子间强烈的库仑相互作用影响,导致脱溶动力学迟缓及电极–电解液界面电荷转移能垒升高。镁的电化学沉积包括Mg2+脱溶、电子转移生成中性原子、表面扩散及成核聚集等基本步骤。尽管镁–镁强金属键及较高的成核势垒有效抑制了枝晶生长,使其在理想条件下形成致密平滑的膜状沉积,但在高电流密度下仍可能出现成核不均匀。依据经典异质成核理论,镁沉积初期的成核过程源于体相自由能降低与表面能增加的竞争。体相自由能下降是成核的驱动力,而表面能代价形成了必须克服的能垒。当成核尺寸超过临界半径后,体系自由能转为负值,金属镁生长在热力学上变得有利。进一步分析表明,降低集流体与镁之间的接触角可显著减少成核能垒,因此提升集流体的“亲镁性”被认为是促进均匀成核的重要策略。
镁负极表面固态电解质界面膜(SEI)的形成机制与电解液组分的最低未占分子轨道(LUMO)能级密切相关。低LUMO能级电解液更易发生还原分解,从而诱导SEI生成。然而,不同于锂体系中自发形成且稳定的SEI,镁体系中的SEI往往不均一,离子电导率低,导致界面阻抗在循环中增加。通过引入添加剂、构建分子中间层或表面预处理以调控人工SEI,是当前提升镁负极稳定性的有效方向。针对电解液结构与界面化学的复杂性,研究者综合采用多种谱学、同步辐射表征及理论计算手段进行解析。通过实验表征与理论计算的协同,建立了从分子结构到界面反应的量化关联模型,为镁电解液体系的理性设计和性能优化提供了坚实的理论基础。
4. 镁电池电解液研究进展
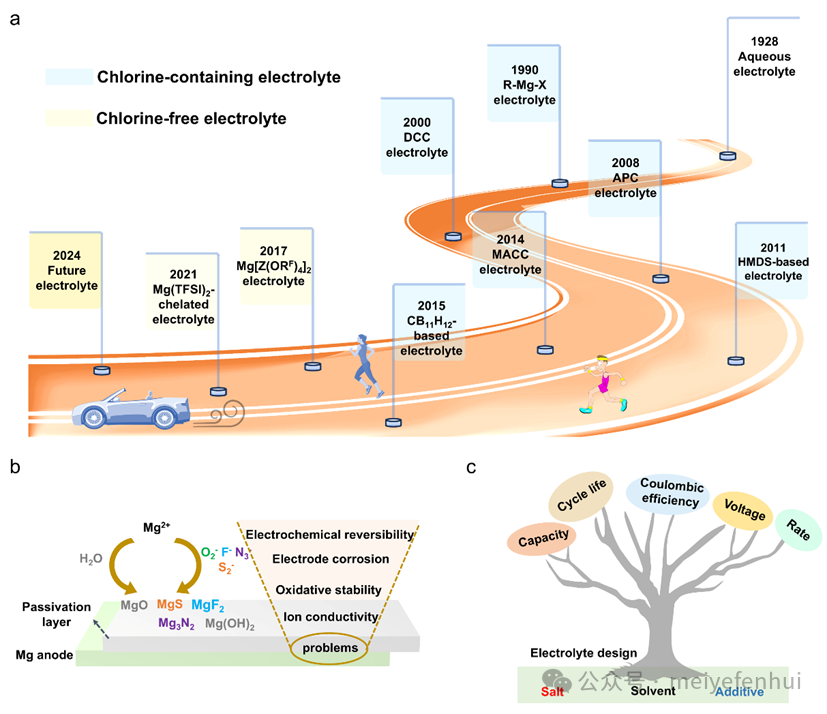
图4. (a)非水系镁电池电解液发展历程的示意图;(b) 电极表面钝化现象的示意图;(c) 电解液对镁电池电化学性能影响机制的示意图。
电解液体系的持续优化对提升镁电池的电化学性能至关重要。非水系镁电解液的发展经历了从含氯体系向无氯体系的演进。早期电解液以熔融MgCl2为代表,虽可实现镁的电化学沉积,但在有机溶剂中溶解度极低,电化学活性有限。随后,人们发现有机化学中常用的Grignard试剂可作为镁电池电解液,实现可逆的镁沉积/剥离,但其对空气与水分极为敏感,稳定性差。通过引入路易斯酸(如卤代铝酸盐)调控溶液配位结构,形成了第一代二氯络合物(DCC)电解液,显著拓宽了电化学稳定窗口。之后发展出的全苯基络合物(APC)电解液进一步提升了氧化稳定性与可逆性。
此后,在传统镁盐体系(如Mg(TFSI)2、Mg(HMDS)2、Mg(Otf)2)中添加氯化物(如MgCl2、AlCl3)可显著改善离子电导率与稳定性,奠定了含氯电解液的主流地位。然而,这类体系具有较强腐蚀性和潜在环境风险,制约了其在大规模储能中的应用。相比之下,无氯电解液体系因其更高的安全性与环境友好性而受到重视,常用镁盐包括Mg(TFSI)2、Mg(Otf)2、Mg(HMDS)2和Mg(B(hfip)4)2。其中Mg(TFSI)2是研究最广泛的体系之一,表现出较宽的电化学窗口(可达4.0 V vs. Mg2+/Mg),但TFSI−易与金属镁反应形成钝化膜,从而影响可逆性。引入胺类添加剂可在一定程度上改善溶剂化结构与界面反应,但其亲核性与低氧化稳定性仍为主要挑战。
镁电解液体系的选择取决于金属–电解液相容性、氧化稳定性、离子电导率以及正极反应机制。由于Mg金属活性极高,易与有机物或水反应生成MgO、Mg(OH)2、MgF2等钝化产物,严重阻碍电子与离子传输。此外,Mg2+与溶剂间强配位作用导致高脱溶能垒,限制了界面电荷转移速率与倍率性能。因此,从分子层面精准调控阴离子类型、溶剂结构与功能添加剂,是实现高性能无氯镁电解液体系的核心策略。
5. 镁电池电解液常用盐、溶剂和添加剂

图5.镁电池电解液中具有代表性的盐类、溶剂及添加剂的分子结构示意图。
通过合理设计和组合这些分子,可显著提升镁电池的电化学稳定性与循环性能。
6. 结论与展望
镁电池凭借高能量密度、低成本及优异安全性,展现出作为新一代储能体系的巨大潜力。然而,Mg2+的二价特性导致其高电荷密度和强极化效应,从而引发脱溶剂化困难和界面反应受限,成为制约镁电池性能提升的关键瓶颈。电解液的设计与优化在其中起决定性作用,其须要同时满足镁负极可逆沉积/剥离与高电压正极兼容性。现有研究系统比较了含氯与无氯两类电解液的结构特征与性能差异。含氯电解液具有高离子电导率(≈ 10−3S cm−1)及优异的可逆性(库仑效率>99%),并能有效抑制负极钝化,但其强腐蚀性及潜在环境风险限制了大规模应用。无氯电解液在安全性和稳定性方面表现突出,具备较宽电化学窗口(3.5–4.3 V vs. Mg2+/Mg)和良好的界面兼容性,但仍存在镁沉积可逆性不足与脱溶剂化能垒高等问题。对比两者可发现,含氯体系通过Cl−的桥联效应有效降低脱溶剂化能垒、促进Mg2+迁移,而无氯体系因溶剂化结构紧密虽具有较高的离子导电率,却受界面极化限制,难以实现稳定的长循环性能。未来的电解液研究应在宏观传输性能与界面动力学之间寻求平衡,通过分子层面调控溶剂化结构与离子缔合行为以实现性能最优化。
镁电池电解液的发展需从多维度协同推进:一方面应聚焦新型盐类、溶剂与添加剂体系的设计,探索弱配位阴离子与高稳定溶剂组合,以降低副反应并构筑稳定SEI;另一方面需深化对镁负极电双层结构的理解,通过界面工程调控成核行为与沉积形貌,提升可逆性与循环寿命。引入创新概念如高熵电解液、高浓度电解液,有望突破液态体系在泄漏、挥发及界面阻抗方面的限制。同时,人工智能(AI)与机器学习技术的引入,将通过数据驱动的高通量筛选加速电解液成分优化,揭示盐–溶剂–添加剂间的非线性结构–性能关系,实现从经验到预测的转变。此外,电解液的设计需与不同类型正极材料(包括高电压嵌入型氧化物、转化型硫基正极以及有机正极)相匹配,兼顾氧化稳定性、离子传输和界面兼容性,以最大化能量输出与循环稳定性。总体而言,镁电池的未来发展将依托于电解液分子设计、界面调控与智能化筛选的有机融合,构建高稳定、高效率、环境友好的储能体系,推动其由实验室研究迈向实际应用。


