可充电镁电池(RMBs)代表了一种颇具前景的后锂技术,然而其发展从根本上受到高容量正极中Mg²⁺缓慢的去溶剂化过程以及转化反应不可逆性限制。2026年02月28日,重庆大学/明月湖实验室潘复生院士、徐朝和、瞿佰华、李胜洋团队在Advanced Functional Materials期刊发表题为“Breaking Kinetic Bottlenecks in Rechargeable Magnesium Batteries: Interfacial Sb Doping for Dual-Function Desolvation and Reconversion in CuS Cathodes”的研究论文,李胜洋为论文第一作者,潘复生院士、徐朝和、瞿佰华为论文共同通讯作者。

第一作者:李胜洋
通讯作者:潘复生、徐朝和、瞿佰华
通讯单位:重庆大学
论文DOI:10.1002/adfm.74684
该研究报道了一种针对硫化铜(Sb-CuS)的合理界面锑(Sb)掺杂策略,该策略同时解决了这两个动力学瓶颈。理论计算与实验相结合表明,Sb掺杂不仅创造了电荷重分布区域,显著降低了Mg²⁺的去溶剂化能垒(7.22 eV),而且还参与形成了Mg₃Sb₂纳米合金,该合金在充电过程中催化了MgS的分解。这种协同作用使得高度可逆且快速双向CuS/MgS转化动力学成为可能,并有效抑制了Cu物种向阳极的扩散。因此,Sb-CuS正极在50 mA g⁻¹电流密度下提供了365 mAh g⁻¹的高可逆容量,并具有出色的长期循环稳定性(在1 A g⁻¹下循环800次后容量为107 mAh g⁻¹,对应91%的容量保持率)。此外,功能性软包电池证明了其实用可行性。该研究阐明了一种适用于转化型硫族化合物正极的通用界面工程范式,为获得高性能多价态离子电池提供了一条关键途径。
可充电镁电池(RMBs)由于镁的天然丰度、更高安全性和高理论体积容量(3833 mAh cm⁻³),作为后锂储能的有前景候选者而备受关注。尽管在电解液和负极设计方面取得了实质性进展,但对高性能正极材料的探索仍然是一项关键瓶颈。尽管嵌入型正极,如Chevrel相Mo₆S₈和钒氧化物,提供了合理的工作电压和结构稳定性,但其实际容量受到刚性晶体框架的内在限制(<150 mAh g⁻¹),从而阻碍了其在RMBs中的广泛应用。因此,开发高容量正极材料是推动RMB技术走向实际应用的一项紧迫且必要的挑战。
转化型正极材料为高能量密度镁电池提供了一条有前景的途径,其中硫基化合物因其"软"阴离子晶格而成为特别有吸引力的候选材料。这种结构特征有效减轻了与Mg²⁺离子的强静电相互作用,从而促进了离子扩散并增强了反应动力学。在这些材料中,六方硫化铜(CuS)因其高理论可逆容量(560 mAh g⁻¹)和优异的本征电子电导率(~10⁻³S cm⁻¹)而引起了极大的兴趣。然而,CuS在RMBs中的电化学性能仍然受到两个主要动力学和可逆性挑战的阻碍。第一个限制源于Mg²⁺在电极-电解液界面处的高去溶剂化能垒。二价Mg²⁺与常规电解液中溶剂分子之间的强库仑相互作用导致缓慢的去溶剂化动力学,从而产生显著的极化和较差的倍率性能。第二个关键问题在于转化反应的不可逆性。最终放电产物硫化镁(MgS)表现出高电化学惰性,并且在充电过程中具有极慢的再转化动力学,这导致了快速容量衰减和有限循环寿命。在反复循环过程中,结构逐渐坍塌和活性硫的不可逆损失进一步加剧了这些问题。
为了应对这些挑战,人们设计了多种策略来提高CuS正极的电化学性能,主要通过结构工程,例如纳米化以缩短离子传输距离、优化电极结构以及与导电碳基体复合。虽然这些方法可以在一定程度上提高容量保持率,但其通常无法从根本上改变CuS的本征电子结构,也无法减轻Mg²⁺与主体晶格之间的强库仑相互作用。相比之下,界面掺杂工程最近作为一种精确调控硫化物基正极中电极-电解液界面的方法而出现。与通过改变晶格内部的本征电子结构和离子扩散路径来改善材料电化学性能的体相掺杂不同,界面掺杂策略选择性地修饰关键异质界面处的局部电子环境,为调控界面电荷转移和离子去溶剂化动力学提供了一条有效途径。
例如,He等人开发了一种Fe³⁺掺杂的尖晶石氧化物(CoMnFe)中间层,其中Fe³⁺调节了Co³⁺和Mn³⁺的电子结构,以促进目标溶剂的捕获并促进去溶剂化。类似地,Zhang等人采用Cu掺杂CeO₂来调整界面电子云分布,从而增强了对Zn²⁺的化学亲和力[25]。尽管在其他电池系统中取得了这些进展,但界面掺杂在调控Mg²⁺去溶剂化行为方面的作用在RMBs中仍未得到充分探索。此外,RMBs中现有的掺杂研究主要集中于增强放电过程,而对于控制从MgS到CuS的关键再转化反应的充电过程仍知之甚少。因此,关键挑战在于合理设计多位点协同的界面掺杂结构,并从根本上阐明其在放电和充电过程中的双向工作机制,最终实现具有高可逆性和快速反应动力学的CuS正极。
在此,该研究提出了一种合理的界面Sb掺杂策略,以同时应对RMBs中CuS正极面临的Mg²⁺去溶剂化缓慢和转化反应不可逆双重挑战。通过阳离子交换方法合成了界面Sb富集的CuS纳米片。研究表明,表面Sb掺杂诱导了电荷重新分布的区域,将Mg²⁺的去溶剂化能垒从9.65 eV降低到7.22 eV。同时,原位形成的Mg₃Sb₂纳米合金催化促进了MgS的分解,其能垒降低至0.91 eV。这种双功能机制不仅确保了具有平衡双向动力学的高度可逆CuS/MgS转化,而且有效抑制了Cu和S物种的溶解。受益于这些协同效应,水热合成的Sb掺杂CuS正极展现了卓越的镁存储性能,其优异的倍率性能和循环稳定性大幅超越了传统的CuS材料。该研究为获得高性能CuS正极提供了一种有效的界面工程方法,并为克服多价态离子电池中转化型电极的动力学限制建立了基本设计原则。
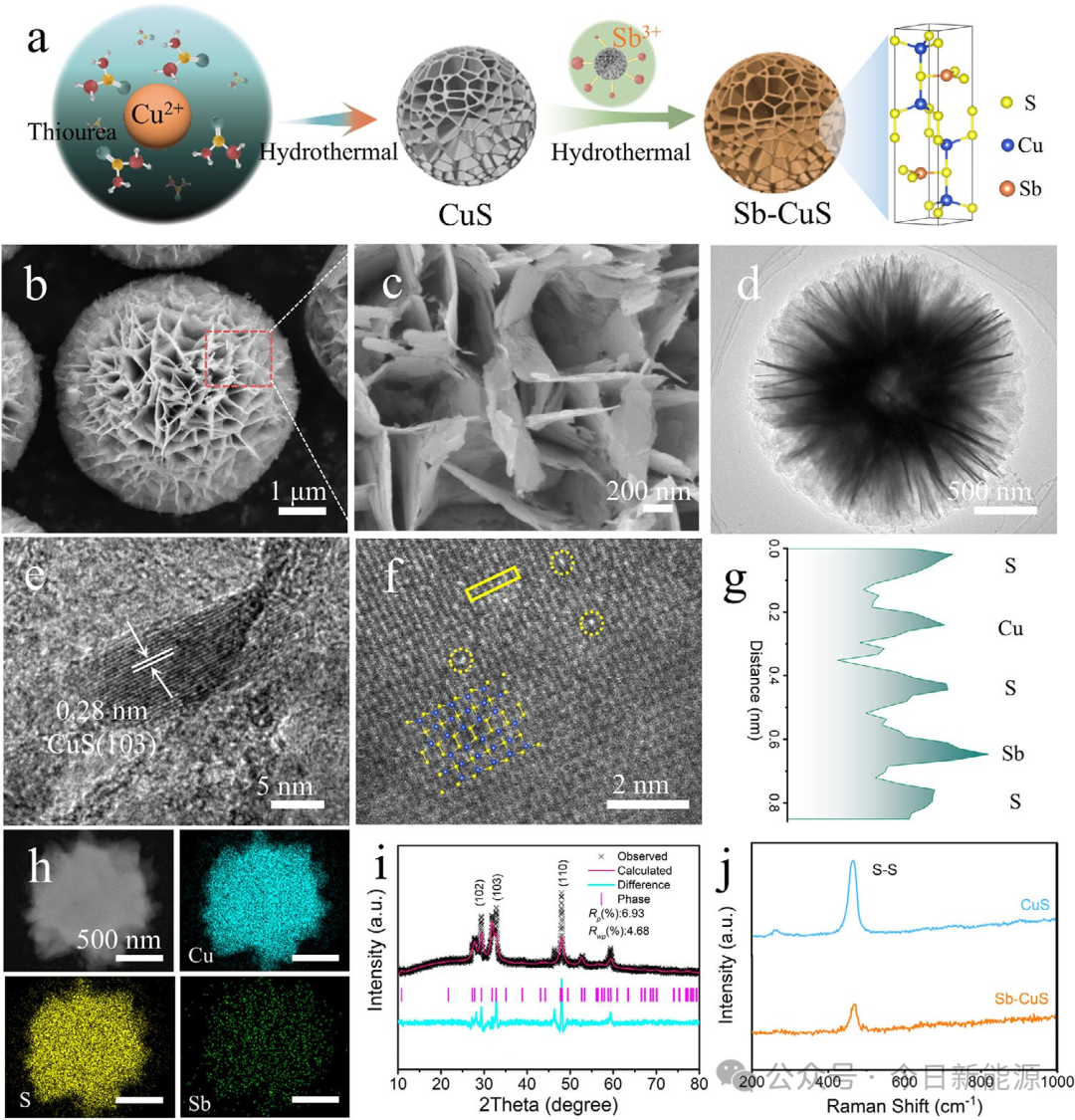
图1 (a) Sb-CuS制备过程示意图。(b, c) Sb-CuS的SEM图像。(d, e) Sb-CuS的TEM和HRTEM图像。(f, g) Sb-CuS的STEM图像以及沿选定区域的强度分布图。(h) 元素分布图。(i) XRD精修图谱。(j) 拉曼光谱。
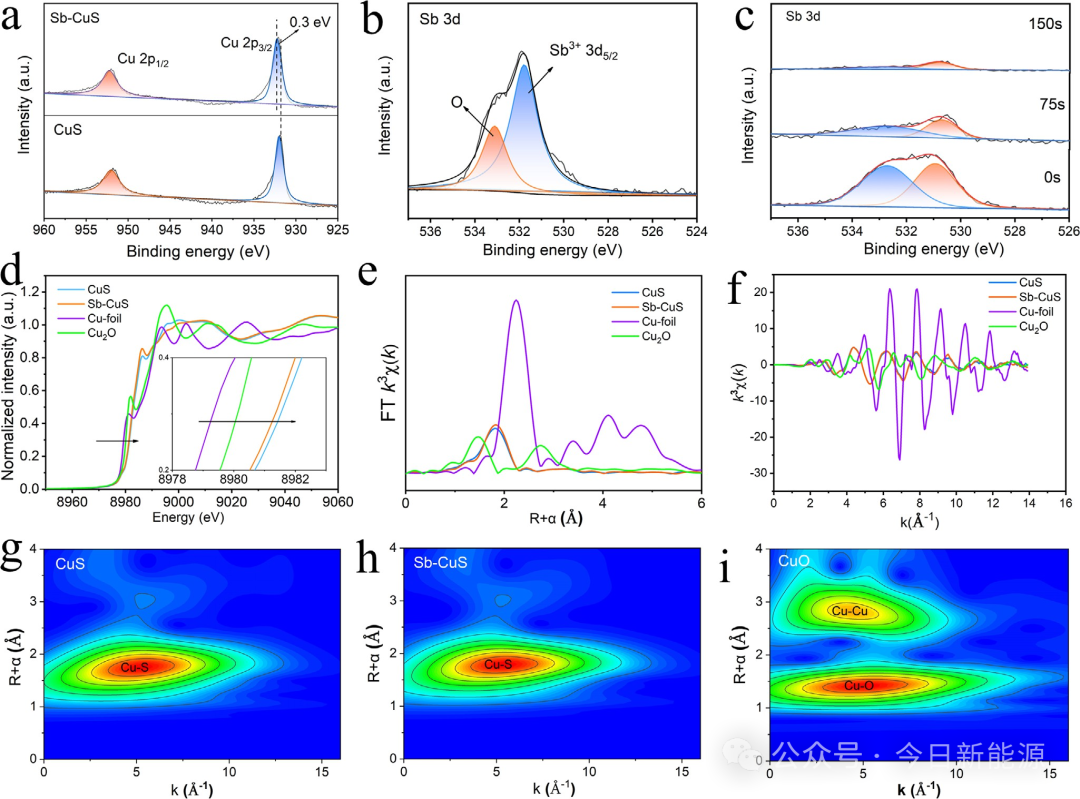
图2 (a, b) CuS和Sb-CuS中Cu 2p的高分辨率XPS谱图以及Sb-CuS中Sb 3d的高分辨率XPS谱图。(c) Sb-CuS的XPS深度刻蚀。(d) Sb-CuS和CuS的Cu K-edge XANES谱图。(e) 傅里叶变换(FT)k³加权χ(k)-函数的R空间图。(f) k空间和R空间的EXAFS拟合谱图。(g–i) 小波变换(WT)EXAFS图。
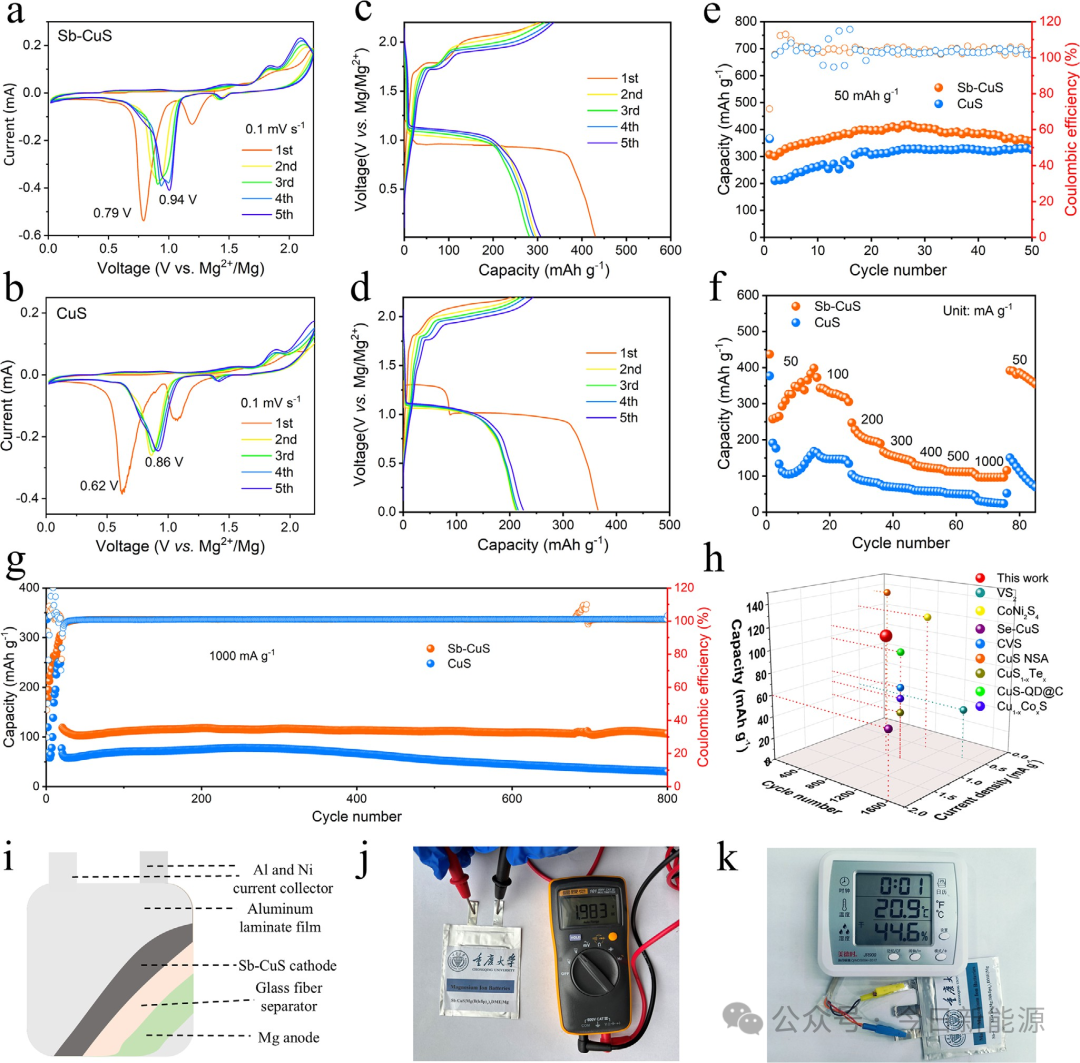
图3 (a, b) Sb-CuS和CuS的CV曲线图。(c, d) Sb-CuS和CuS的GCD曲线。(e) Sb-CuS和CuS在50 mA g⁻¹下的循环性能。(f) 倍率性能。(g) Sb-CuS和CuS在1000 mA g⁻¹下的长循环性能。(h) Sb-CuS与先前报道的其他代表性正极的电化学性能比较。(i) 软包电池结构示意图。(j) Sb-CuS||Mg软包电池的开路电位。(k) 由Sb-CuS||Mg软包电池供电的电子时钟。
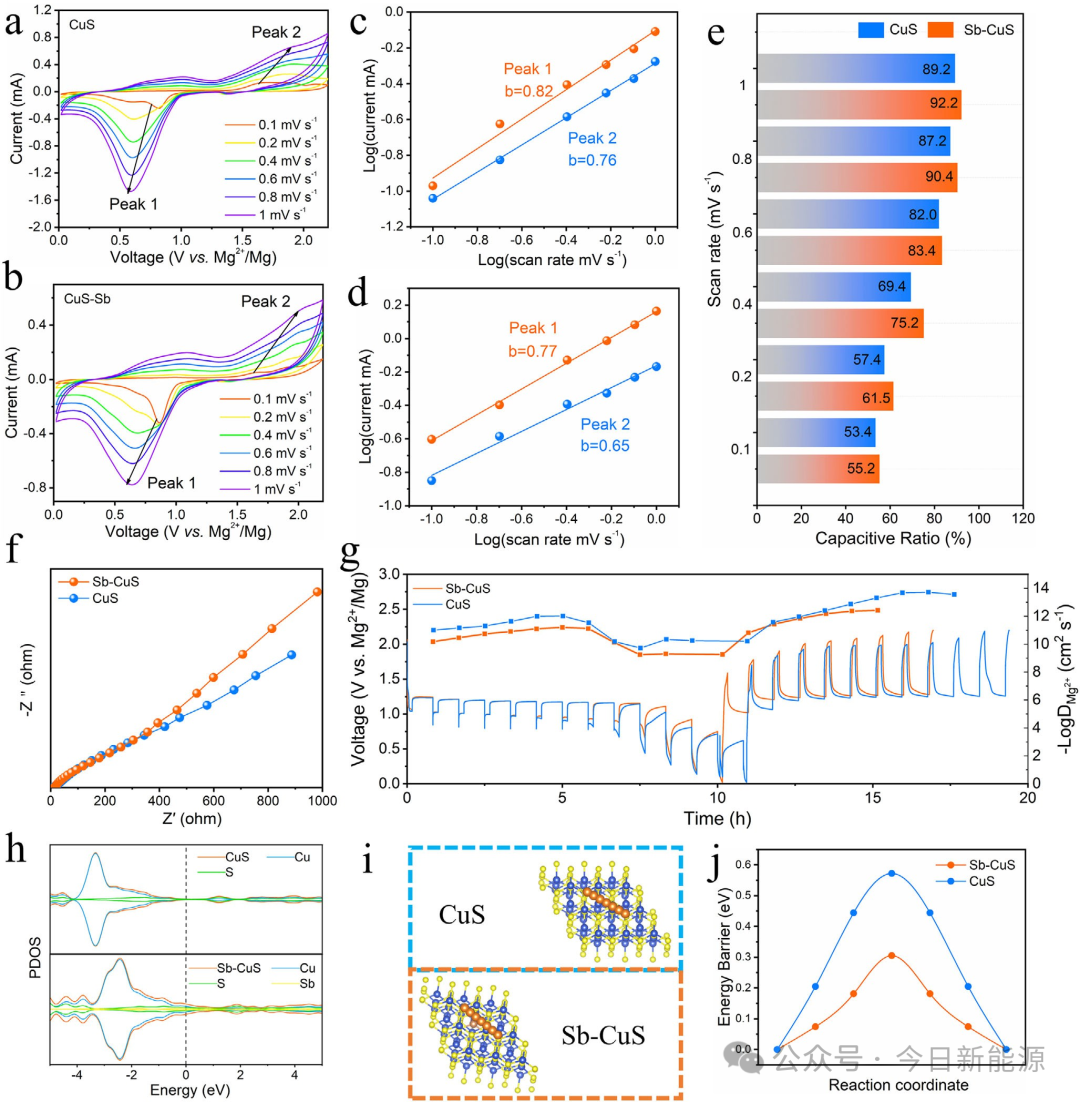
图4 (a, b) Sb-CuS和CuS在不同扫描速率下的CV曲线。(c, d) Sb-CuS和CuS的log(i)与log(v)关系图。(e) 不同扫描速率下电容贡献的百分比。(f) Sb-CuS和CuS的Nyquist图。(g) 由GITT计算得出的Mg²⁺扩散系数。(h) CuS和Sb-CuS的态密度(DOS)。(i, j) CuS和Sb-CuS中Mg²⁺的扩散能垒。
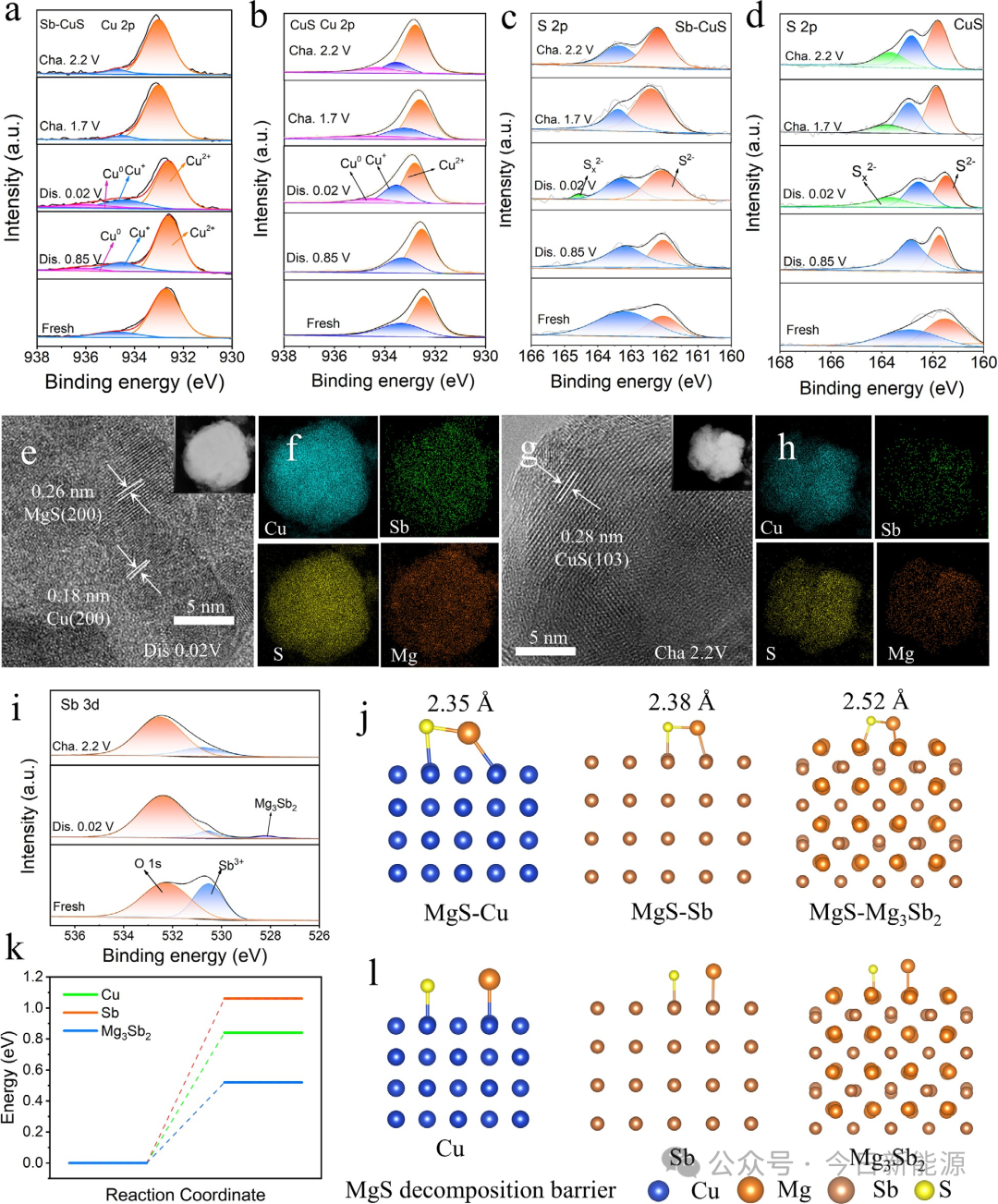
图5 (a, b) Sb-CuS和CuS中Cu 2p的非原位高分辨率XPS谱图。(c, d) Sb-CuS和CuS中S 2p的非原位高分辨率XPS谱图。(e-h) Sb-CuS在第一次放电和第一次充电后的HRTEM图像及相应的EDS元素分布图;插图部分显示了电极的整体形貌。(i) Sb 3d的非原位高分辨率XPS谱图。(j) 吸附在Cu、Sb和Mg₃Sb₂基底上的Mg-S键长。(k, l) 在Cu、Sb和Mg₃Sb₂基底上的分解能垒和MgS分解过程。
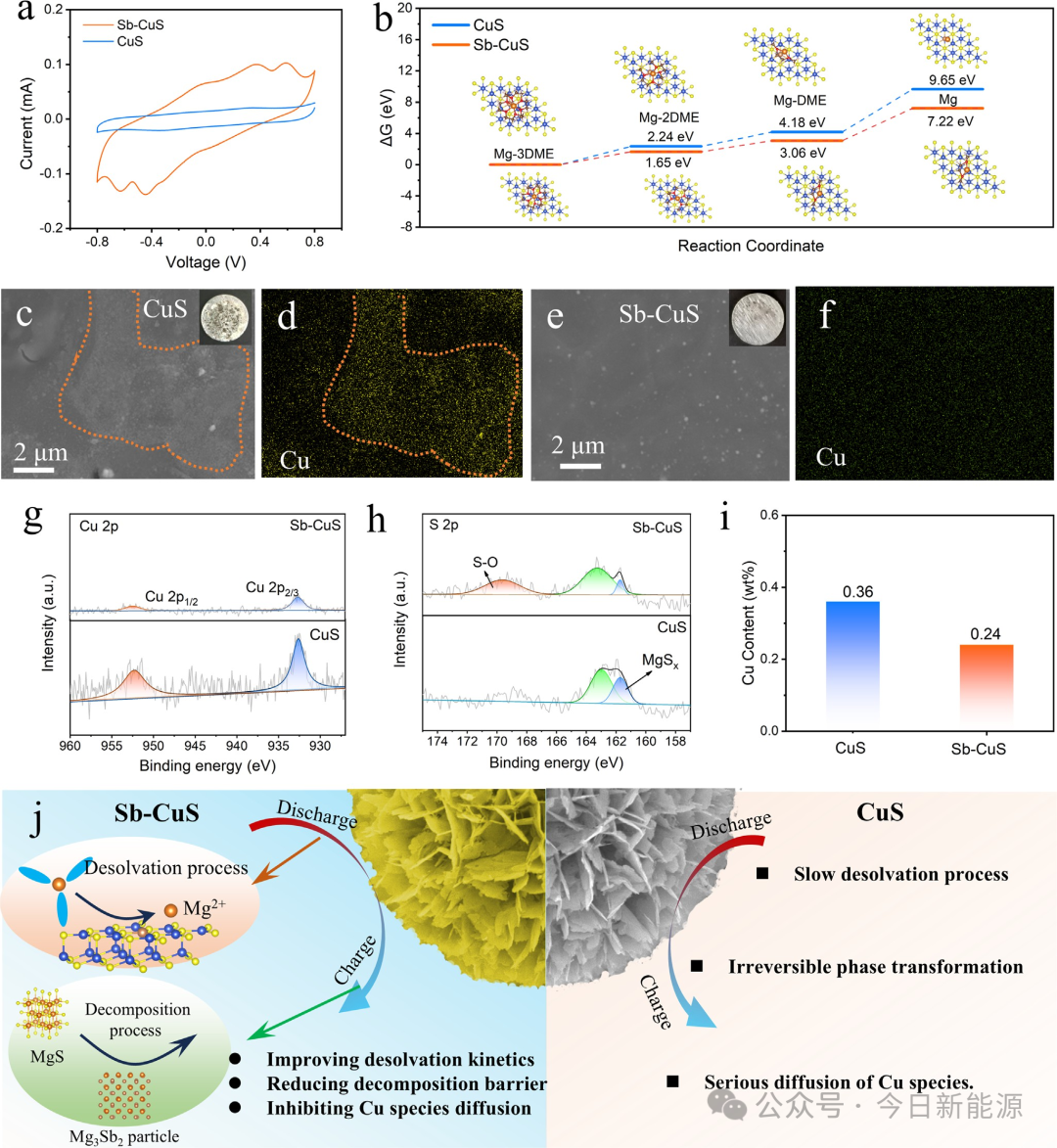
图6 (a) 基于Sb-CuS和CuS对称电池的CV曲线。(b) Mg²⁺溶剂团的去溶剂化步骤以及在Sb-CuS和CuS上的能量变化。(c-f) 使用不同正极循环50次后镁负极的SEM图像,以及相应的Cu元素分布图,插图为镁负极的照片。(g, h) 镁负极上Cu 2p和S 2p的XPS谱图。(i) 镁负极上的Cu含量。(j) Sb掺杂在增强CuS电化学性能中的双功能机理示意图。
总之,该研究证实了在CuS正极中进行合理的界面Sb掺杂,可以同时解决可充电镁电池中缓慢Mg²⁺去溶剂化和不可逆转化这两个动力学限制。通过一系列全面表征和密度泛函理论DFT计算,阐明Sb诱导的电荷再分布区域将Mg²⁺去溶剂化能垒降低至7.22eV,同时原位形成的Mg₃Sb₂纳米合金催化促进MgS分解,其能垒降低至0.91eV。这种双功能策略确保了高度可逆的相转变并加速了其动力学过程,同时有效抑制了活性材料的溶解。因此,Sb-CuS正极在50mA g⁻¹电流密度下提供了365mAh g⁻¹的高可逆容量,并具有优异的长期循环稳定性,在1A g⁻¹下循环800次后仍保持107mAh g⁻¹的容量。该研究还在一个实用软包电池构型中进一步证明了其可行性。从根本上说,该研究提供了一种通用界面工程范式,可扩展到其他转化型硫族化合物正极,为高性能多价态电池开辟了一条有力途径。










